@MSFTResearch: Talos was built to help resolve a major bottleneck in genomic medicine: human review time. The open-source system recov…
Summary
Talos is an open-source tool for automated, iterative genomic reanalysis that resolves the bottleneck of human review time, recovering 90% of diagnoses while surfacing only 1.3 candidate variants per patient.
View Cached Full Text
Cached at: 06/24/26, 04:04 PM
Talos was built to help resolve a major bottleneck in genomic medicine: human review time. The open-source system recovered 90% of in-scope diagnoses while surfacing just 1.3 candidate variants per patient for expert review. https://t.co/tBnSoU1aXy https://t.co/Sd9HSVOug3
Talos brings continuous genomic reanalysis to nearly 5,000 unsolved cases
Source: https://www.microsoft.com/en-us/research/blog/talos-scaling-rare-disease-diagnosis-with-automated-iterative-genomic-reanalysis/
 ## At a glance
## At a glance
- Talos is an open-source tool for automated, iterative reanalysis of genomic data in rare disease. It efficiently re-examines stored sequencing data as scientific knowledge evolves and flags variants with newly actionable evidence.
- Talos is tuned for a low false-positive rate: across a validation set of nearly 1,100 patients, it recovered 90% of in-scope diagnoses while flagging only 1.3 candidate variants per patient for expert review. This is essential to making reanalysis sustainable at scale.
- Deployed across a prospective cohort of almost 5,000 undiagnosed patients, Talos delivered 241 new diagnoses (5.1% additional yield). An average of only 32 days passed between supporting evidence becoming public and the resultant diagnosis.
- On monthly iterative cycles, analysts only needed to review one new variant per 200 patients, demonstrating that frequent, systematic reanalysis can be run sustainably.
Why genome reanalysis matters
Genomic testing has transformed the diagnosis of rare disease, but even with this advancement, more than half of patients remain undiagnosed after their first test. This is because our knowledge of the genome is still incomplete. Researchers are learning more every day about the function of specific genes and how they relate to disease.
However, unlike most diagnostic investigations, genomic data has a unique property: it can be stored and reexamined indefinitely. Because our understanding of the genome improves constantly, simply rerunning the analysis later can yield a diagnosis that was impossible to make the first time. This is because there are hundreds of new gene–disease associations and thousands of new variant classifications reported every year.
Reanalysis of the genomes of undiagnosed patients is the solution; a meta-analysis of nearly 9,500 undiagnosed patients found that reanalysis lifted diagnostic yield by about 10% over roughly two years. However, the problem is that reanalysis today is overwhelmingly manual. It depends on motivated clinicians, scarce laboratory staff, and inconsistent reimbursement, so the vast majority of stored genomes are never revisited and the data keep accumulating. Automation has long been proposed as the answer, but the developers of automated machinery must navigate hard trade-offs between sensitivity, specificity, how many candidate variants a human must review, and how often the analysis is rerun.
Talos(opens in new tab), developed through a collaboration spanning the Centre for Population Genomics, Australian Genomics, the Broad Institute, and Microsoft, was built to resolve those trade-offs and to demonstrate, at international scale, that systematic reanalysis is both feasible and valuable. We have recently published ajournal article(opens in new tab)detailing how Talos functions and evaluating its performance on multiple rare disease cohorts.
How Talos works
Talos re-interprets a patient’s existing variant calls against the latest community knowledge each time it runs. It draws on two continuously updated public resources:PanelApp Australia(opens in new tab)for gene–disease relationships and modes of inheritance, andClinVar(opens in new tab)for variant-level pathogenicity. It then applies a variant-prioritization algorithm designed to surface variants most likely to meet ACMG/AMP criteria for clinical reporting.
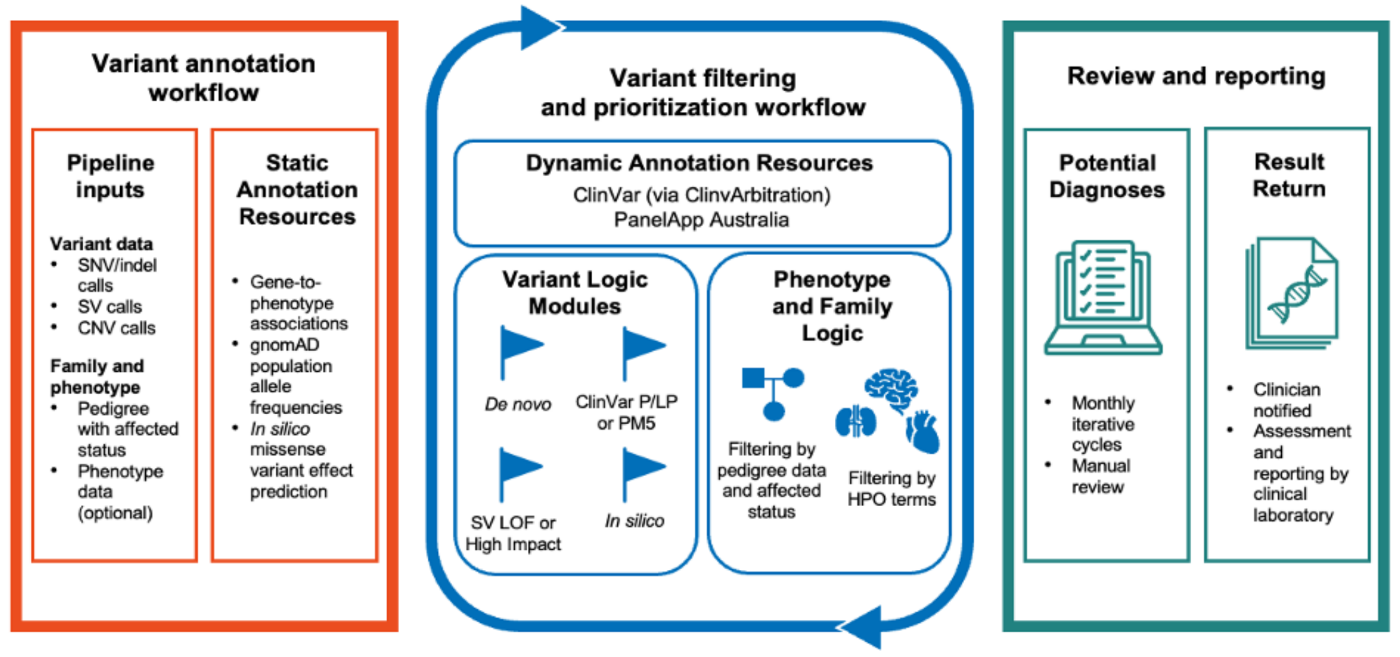 ****Figure 1 – Talos overview.****Talos operates in multiple stages, first collecting unchanging information about genetic variants and the patients who possess them, then applying up to date knowledge to filter and prioritize variants that are likely to be clinically relevant, then finally surfacing those variants to clinicians alongside supporting evidence.**The pipeline uses newly discovered information to tag and filter variants, then refines the candidate set using family structure (for example, mode of inheritance and de novo status) and, when available, the patient’s phenotype. Talos can be used to interpret single-nucleotide variants, small insertions/deletions, copy number variants, and large structural variants from exome or genome data.
****Figure 1 – Talos overview.****Talos operates in multiple stages, first collecting unchanging information about genetic variants and the patients who possess them, then applying up to date knowledge to filter and prioritize variants that are likely to be clinically relevant, then finally surfacing those variants to clinicians alongside supporting evidence.**The pipeline uses newly discovered information to tag and filter variants, then refines the candidate set using family structure (for example, mode of inheritance and de novo status) and, when available, the patient’s phenotype. Talos can be used to interpret single-nucleotide variants, small insertions/deletions, copy number variants, and large structural variants from exome or genome data.
Two design choices distinguish Talos. First, it is deliberately conservative, optimized to return a small set of high confidence variants rather than a long ranked list, because in real-world genomic reanalysis the limiting factor is human review time, not algorithmic recall. Second, on repeat runs, Talos returns only variants whose supporting evidence has changed since the previous cycle, allowing clinicians to focus exclusively on findings that aregenuinely new.
Validated against expert manual analysis
We benchmarked Talos on two independent cohorts that had already undergone careful manual analysis: the Australian Acute Care Genomics (ACG) cohort of critically ill infants and children, and the U.S.-based Rare Genomes Project (RGP) cohort of families with prior uninformative testing. This included 1,089 probands in total.
On ACG trios, Talos recovered 90% of in-scope diagnoses while returning a median of just 1.3 candidate variants per family. The diagnoses it missed were largely a direct consequence of its conservative strategy, for example, recessive variants lacking ClinVar support that human analysts had classified usingtransconfiguration or functional studies.
Crucially, Talos held the same operating point on the very different RGP cohort, agroup of families who had previously had uninformative clinical testing, with probands ranging up to 82 years of age. On RGP trios, it recovered 87% of in-scope diagnoses (47 of 54) at a median of 1.3 candidate variants per trio, showing generalizability across cohorts.
We then benchmarked head-to-head against Exomiser, a widely used prioritization tool. Talos matched its overall sensitivity for small variants, but at a very different operating point: Exomiser ranks and returns a broad list, while Talos returns a short, highly specific one. In a paired comparison, the two tools were statistically indistinguishable when all of Exomiser’s ranked variants were reviewed, but Talos came out significantly ahead once review was limited to a realistic budget—the top five (p = 0.017) or top one (p < 0.0001) ranked variants. Notably, the two tools surfaceddifferentvariants, so they are complementary and should ideally be used together in diagnostic workflows.

Azure AI Foundry Labs
Get a glimpse of potential future directions for AI, with these experimental technologies from Microsoft Research.
Deployed on an international scale
The experiment we were most excited about was a tested-but-undiagnosed cohort of 4,735 individuals, drawn from Australian Genomics research studies and a single diagnostic laboratory. Most patients were singletons with neurodevelopmental, cardiac, renal, and/or neurological indications.
Talos produced 241 new diagnoses in 238 individuals—a 5.1% additional yield, with every single likely-causative variant subsequently confirmed as pathogenic or likely pathogenic by accredited labs.
The sources of those diagnoses illustrate why reanalysis is such a powerful paradigm:
- 32% came from new gene–disease relationships discovered since the original test,
- 22% came from new variant-level evidence (reclassifications), and
- 45% came from improved filtering and analysis—including variant types such as CNVs and structural variants not examined originally, phenotype filters that had been set too narrowly, and other sources.
Yield was consistent across clinical areas (roughly 5–6% for neurodevelopmental, cardiac, and renal indications) but thereasonsdiffered: new gene associations and CNVs dominated neurodevelopmental diagnoses, while variant reclassification drove most cardiac ones. Genome data outperformed exome (6.1% vs 4.8%), partly by reaching non-coding diagnoses such asRNU4-2and a deep-intronicMRPL39variant. A recurring theme was the lag in conventional knowledge bases: 59% of the new gene–disease diagnoses were not yet curated in OMIM at the time of reanalysis, underscoring the value of drawing on a rapidly updated resource like PanelApp Australia.
From a one-off event to a continuous program
We then ran Talos for 29 monthly iterative cycles. Most diagnoses (92%) came on a cohort’s first pass, but the iterative design proved its value on two fronts. First, it demonstrated the scalability of ongoing reanalysis: because later cycles return only newly actionable evidence, they surfaced an average of just one variant per 200 cases over the program. Second, it showed how quickly we can move from scientific discovery to diagnosis: on average just 32 days passed between new knowledge appearing in a public database and a patient receiving a diagnosis, with the fastest case turning around in a single day. Figure 2 provides timelines for three example patients showing how continual reanalysis can bring answers to families within weeks of new scientific findings. The whole pipeline is cheap enough to run continuously: annotating 1,000 genomes cost about $11, and a monthly reanalysis pass ran for a few cents per cohort.
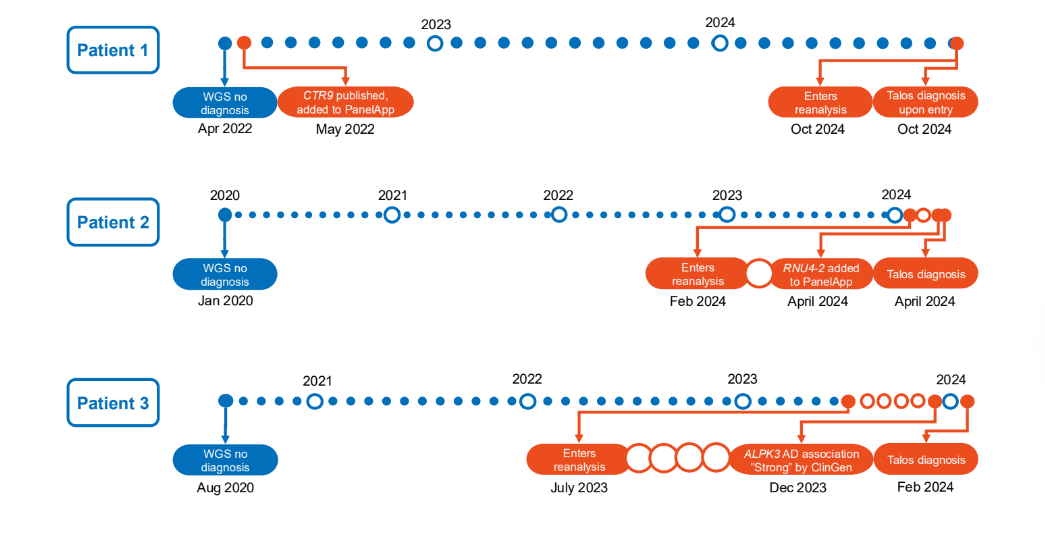 Figure 2 – Diagnostic odyssey for three example patients. Each patient spent years after genetic sequencing waiting for a diagnosis. For Patient 1, the scientific discovery enabling their diagnosis happened one month after their testing, but no diagnosis was made until the first time their genetic data was reanalyzed using Talos. For patients 2 and 3, diagnoses were made within a month of the relevant scientific findings because the patients were already in the reanalysis pipeline.## Looking ahead
Figure 2 – Diagnostic odyssey for three example patients. Each patient spent years after genetic sequencing waiting for a diagnosis. For Patient 1, the scientific discovery enabling their diagnosis happened one month after their testing, but no diagnosis was made until the first time their genetic data was reanalyzed using Talos. For patients 2 and 3, diagnoses were made within a month of the relevant scientific findings because the patients were already in the reanalysis pipeline.## Looking ahead
Talos reframes genomic reanalysis from a rare, labor-intensive event into a continuous, automated program that can keep pace with the science. By optimizing for specificity, it respects the real bottleneck of expert reviewer time, and by drawing on openly shared, frequently updated resources like PanelApp Australia and ClinVar, it turns the global community’s accumulating knowledge into diagnoses for individual patients, often within weeks.
We believe we’ve established a foundational capability, and we’re excited to see how the community builds on it. In particular, as more advanced AI models for understanding and predicting the consequences of genetic variation become available, we’re looking forward to leveraging them in the reanalysis of unsolved rare disease cases.
Talos is open source and straightforward to deploy in cloud environments like Azure. Our results offer a practical blueprint for health systems aiming to deliver frequent, scalable reanalysis to the many patients still searching for diagnoses.
Similar Articles
@MSFTResearch: 30x faster analytics, GPU kernels generated automatically from SQL, AI matched to lab-grown tumor models for cancer tre…
Microsoft Research highlights multiple advances including 30x faster analytics with CoddSpeed, AI wildlife re-identification, and LLMs that learn across tasks without retraining in the latest Research Focus newsletter.
@OpenAI: Rare disease diagnosis is challenging, as sequencing can surface millions of variants, and medical knowledge changes co…
OpenAI highlights how o3 Deep Research can aid rare disease diagnosis by integrating clinical features, inheritance patterns, variant evidence, and scientific literature into actionable hypotheses for specialists.
@SethSHowes: I just sequenced a human genome to 30× coverage entirely at home. As far as I know, this is the first time this has bee…
A researcher sequenced a human genome to 30× coverage entirely at home using an Oxford Nanopore P2 Solo, marking the first time this has been done outside a lab. He attributed his success to AI helping solve technical problems across software, hardware, and molecular biology.
Turning scattered evidence into discovery decisions for life sciences
OpenAI’s new life-science model “GPT-Rosalind” inside Codex autonomously ranks asthma drug targets by orchestrating specialist sub-agents that merge genetics, transcriptomics, safety and IP data into a single evidence-backed decision.
Using AI to help physicians diagnose rare genetic diseases affecting children
Researchers from Boston Children's Hospital, Harvard, and OpenAI used the OpenAI o3 Deep Research reasoning model to reanalyze 376 unsolved rare disease cases, leading to diagnoses in 18 additional cases (4.8% yield) after expert review and clinical confirmation. The study, published in NEJM AI, demonstrates how AI-assisted workflows can help experts revisit difficult cases as scientific knowledge evolves.